An improved high-resolution method for the in silico detection of EMS-induced mutations in sorghum mutant populations
The accurate detection of bona fide induced mutations among sequenced mutants is critical for gene function discovery. In the era of high-throughput phenotyping, the availability of high-quality mutant resources could have an enormous impact on crop plant breeding programs and improve our understanding of the molecular mechanisms associated with highly desired agronomical traits.
In 2018, the Purdue University sorghum gene function discovery team published a high-quality genetic variant resource derived from low-coverage next-generation sequencing of the M3 generation of an EMS-mutagenized sorghum population (Addo-Quaye et al. 2018). This genetic resource consists of 1,753,403 SNPs including 1,275,872 homozygous SNPs, of which 98% are G:C to A:T substitutions. The subtraction of common DNA variants (Uchida et al. 2011) proved to be critical for uncovering widespread likely false-positive SNPs that had been assigned high-confidence variant-call quality scores.
Addo-Quaye et al improved the SNPs prediction methodology by incorporating identification of false-negative SNPs. They revised the SNP filtering methodology by initially applying common variant subtraction and retaining only SNPs that were not shared among the individuals in the sequenced mutant population. Using the retained SNPs, they performed an empirical determination of the SNP variant-call quality-score threshold for SNP filtering by plotting G:C to A:T percentages versus SNP variant-call quality scores. Using the widely known EMS mutation spectrum as a benchmark, they selected the minimum SNP quality-score threshold (Q=12 for SAMtools; Q=28 for GATK) at which the calculated mutation spectrum of retained SNPs shows evidence of EMS action. This high-resolution approach revealed widespread likely false negatives (~1.5 million SNPs), thereby improving their SNP prediction accuracy. They applied this new approach to both SAMtools- and GATK-based SNP analyses and obtained about 90% (> 3 million SNPs) concordance between the two prediction methods. Importantly, 98% of the final SAMtools SNP predictions overlapped with the GATK predictions. Comparing the SAMtools-based results to the previous analysis (in which 95% of the total number of SNPs were G:C to A:T substitutions) revealed that 96% of sorghum SNPs (3,141,908 out of 3,274,606 SNPs) in the new analysis were G:C to A:T substitutions.
More recently, the authors have also implemented a novel DNA contaminant detection algorithm and used it to successfully uncover further evidence of new false negatives. Their ongoing research work is focused on integrating these newly predicted false negatives, and then annotating the final high-quality sorghum EMS-induced DNA mutations from the Purdue University sorghum gene function discovery project.
“It was a great privilege to contribute to the Purdue sorghum EMS mutant population project. It was enlightening to walk through sorghum fields, observing segregating mutant phenotypes, and then comparing with our predicted candidate genes for the 600 mutants and seeing firsthand that genetics combined with bioinformatics really works. I hope plant breeders and the general plant science community find this mutant resource useful.” – Charles Addo-Quaye, Lewis-Clark State College
SorghumBase example
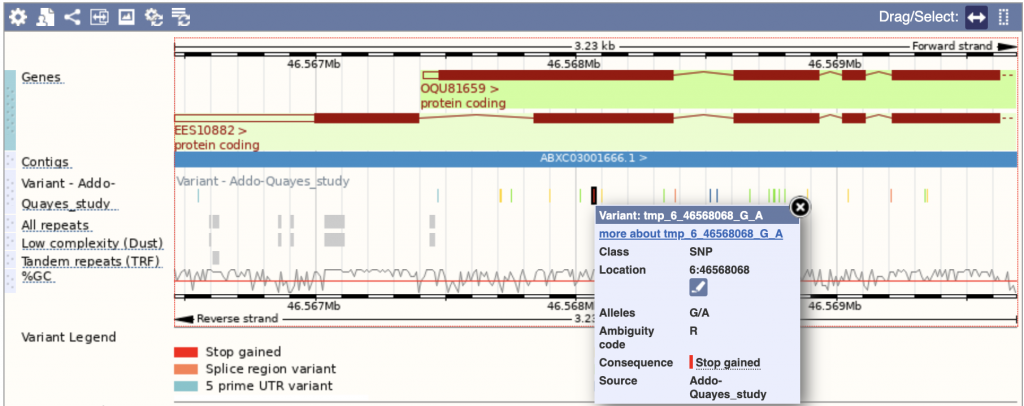
Reference
Addo-Quaye C, Tunistra M, Carraro N, Weil C and Dilkes BP (2018) Whole-Genome Sequence Accuracy Is Improved by Replication in a Population of Mutagenized Sorghum. G3 8(3):1079-1094. DOI: 10.1534/g3.117.300301. Read more
Related Project Websites



